

Was tun bei familiärer Hypercholesterinämie?


Der LDL-Rezeptor: Das für ihn verantwortliche Gen ist bei familiärer Hypercholesterinämie verändert.
© Juan Gärtner - stock.adobe.com
Was ist eine familiäre Hypercholesterinämie?
Die familiäre Hypercholesterinämie ist eine erblich bedingte Störung des Fettstoffwechsels. Der Spiegel eines bestimmten Blutfetts – LDL-Cholesterin – steigt auf über 310 mg/dl an, teilweise auf das Zwei- bis Zehnfache des Normalwertes.

Chylomikronen, LDL, HDL: Cholesterin wird im Körper auf unterschiedliche Weise transportiert.
© W&B/Dr. Ulrike Möhle
Ursache: Veränderung des LDL-Rezeptor-Gens
Die Ursache für die familiäre Hypercholesterinämie ist in den meisten Fällen eine Veränderung (Mutation) des LDL-Rezeptor-Gens – also der Erbinformation, die den "Bauplan" für den LDL-Rezeptor enthält. Dieser LDL-Rezeptor ist eine wichtige Voraussetzung dafür, dass LDL-Cholesterin aus dem Blut in die Zellen gelangen kann. Bei der familiären Hypercholesterinämie funktioniert dieser Mechanismus aber nicht richtig, so dass LDL-Cholesterin nicht oder nur teilweise in die Zellen aufgenommen wird. Die LDL-Cholesterinspiegel im Blut steigen. Daneben sind noch zwei weitere Gene bekannt, deren Veränderungen zu einer familiären Hypercholesterinämie führen (ApoB-, PCSK9-Gen).
Die genetisch bedingte Funktionsstörung der LDL-Rezeptoren kann verschieden stark ausgeprägt sein: Patienten sind besonders schwer betroffen, wenn überhaupt keine Rezeptoren auf den Zellen ausgebildet sind. In anderen Varianten der Erkrankung sind zwar Rezeptoren ausgebildet, diese sind aber unfähig, LDL-Cholesterin zu binden. Oder die Störung besteht darin, dass der Komplex aus LDL und dem LDL-Rezeptor nicht in die Zelle aufgenommen werden kann. Dadurch lagert sich das LDL-Cholesterin vermehrt in verschiedenen Geweben ab, insbesondere den Wänden der Herzkranzgefäße, der Halsschlagadern oder der Hauptschlagader (Aorta).
Heterozygot- und homozygot vererbte familiäre Hypercholesterinämie
Die familiäre Hypercholesterinämie wird autosomal-dominant vererbt. Das bedeutet: Bereits ein entsprechend verändertes Gen (entweder von Vater oder Mutter) reicht aus, um zu erkranken.
Besitzt der Patient ein verändertes und ein gesundes Gen, dann liegt die heterozygote (ungleicherbige) Form der familiären Hypercholesterinämie vor. Die heterozygote Form kommt bei einem von 300 bis 500 Menschen vor. Sie zählt zu den häufigsten angeborenen Stoffwechselerkrankungen des Menschen. Ohne Behandlung treten Beschwerden häufig schon ab dem 30. Lebensjahr auf.
In seltenen Fällen vererben sowohl die Mutter als auch der Vater ihrem Kind ein verändertes Gen. Es besitzt also zwei veränderte Gene für den LDL-Rezeptor. Diese Form der Vererbung heißt homozygot (reinerbig). Diese Variante der familiären Hypercholesterinämie geht mit Gesamtcholesterinwerten von über 500 bis 1200 mg/dl (Milligramm pro Deziliter) einher. Das entspricht etwa 12 bis 31 mmol/l (Millimol pro Liter). Es handelt sich um eine sehr schwere Erkrankung. Ohne Therapie überleben die betroffenen Patienten selten das zweite Lebensjahrzehnt. Die homozytoge Form ist mit einer Häufigkeit von 1:1.000.000 seltener als die heterozygote.

Xanthelasmen sind Fettablagerungen an den Lidern.
© stock.adobe/Svetlana
Symptome
Die familiäre Hypercholesterinämie verursacht zunächst keine Symptome oder körperlichen Beschwerden. Das vermehrt im Blut zirkulierende LDL-Cholesterin lagert sich jedoch mit der Zeit an den Gefäßwänden ab und ist damit wesentliche Ursache der „Arterienverkalkung“ (Arteriosklerose). Werden die Gefäße durch die Arteriosklerose eingeengt, entstehen in den betroffenen Organen Durchblutungsstörungen. Schwerwiegende Komplikationen wie zum Beispiel ein Herzinfarkt können die Folge sein.
Äußerlich macht sich die Fettstoffwechselstörung eventuell durch rötlich bis gelbliche Cholesterinablagerungen an bestimmten Stellen der Haut, in Sehnen (Xanthome) und den Augen (Xanthelasmen, Arcus corneae) bemerkbar.

Die Verdachtsdiagnose "familiäre Hypercholesterinämie" stellt der Arzt oder die Ärztin, wenn er oder sie bei seinem Patienten oder seiner Patientin hohe Cholesterinwerte (Gesamtcholesterin und LDL-Cholesterin) im Blut feststellt und zusätzlich aus der Familiengeschichte der Person erfährt, dass nähere Verwandte ähnlich hohe Cholesterinwerte aufweisen, schon früh an koronarer Herzkrankheit litten oder am plötzlichen Herztod verstorben sind. "Früh" bedeutet dabei für Männer ein Alter unter 55 Jahren, für Frauen unter 60. Auch Fetteinlagerungen in den Sehnen, sogenannte Sehnenxanthome bei der oder dem Betroffenen selbst oder den Angehörigen wecken den Verdacht auf eine familiäre Hypercholesterinämie.
Wie hoch sind die LDL-Werte bei familiärer Hypercholesterinämie?
Bei der leichteren, heterozygoten Form der Krankheit (siehe Abschnitt Ursache) liegen die LDL-Cholesterinwerte im Blut im Bereich 200 bis 400 mg/dl (Milligramm pro Deziliter), wobei die Triglycerid-Werte in der Regel nicht erhöht sind. Manchmal werden die Werte auch in der Einheit Millimol pro Liter (mmol/l) angeben: 200 bis 400 mg/dl entsprechen etwa 5 bis 10 mmol/l.
Bei der selteneren, schweren, homozygoten Form der Krankheit finden sich Gesamtcholesterinwerte von 500 mg /dl bis 1200 mg/dl (das entspricht etwa 12 bis 31 mmol/l).
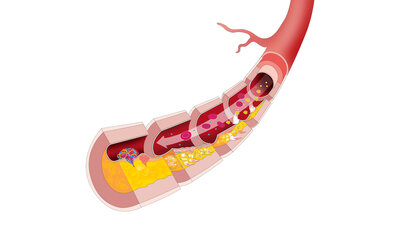

Zur Einschätzung, ob eine familiäre Hypercholesterinämie vorliegen könnte, gibt es auch den sogenannten FH-Score, ein Diagnosewerkzeug für Ärztinnen und Ärzte. Bestätigen lässt sich ein entsprechender Verdacht dann durch einen genetische Untersuchung (DNA-Analyse). Die Kosten dafür übernimmt üblicherweise die Krankenkasse. Wird dabei eine entsprechende Genveränderung nachgewiesen, ist die Diagnose gesichert. Dann sollte die ganze Familie auf das entsprechende Gen untersucht werden. Allerdings findet sich eine Veränderung der drei bisher bekannten Gene für Hypercholesterinämie nur bei etwa 70 Prozent der Personen, bei denen ein Verdacht auf eine familiäre Hypercholesterinämie vorliegt. Möglicherweise sind bei den übrigen mehrere verschiedene Gene verändert oder es liegen bisher noch nicht entdeckte Genveränderungen vor.
Therapie: Was tun bei familiärer Hypercholesterinämie?
Zunächst sollte versucht werden, die Fettstoffwechselstörung durch Ernährungsumstellung und durch Medikamente zu behandeln. Wie hoch der LDL-Wert sein soll, der erreicht werden soll, hängt davon ab, wieviele weitere Risikofaktoren für Herz-Kreislaufkrankheiten bei der betroffenen Person vorliegen. Die Zielwerte für das LDL liegen meist zwischen 50 und 100 mg/dl, je nachdem, ob bereits Folgen einer Arterienverkalkung vorliegen. Sie werden aber individuell mit dem Arzt oder der Ärztin abgestimmt.
Ernährung bei familiärer Hypcherolesterinämie
Für die Ernährungsumstellung gelten folgende Regeln:
- Bei Übergewicht: die Kalorienzufuhr passend zum tatsächlichen Bedarf reduzieren und damit überflüssige Pfunde abbauen.
- Weniger gesättigte (tierische) Fette und Transfette aufnehmen!
- Die Cholesterinzufuhr senken.
- Den Ballaststoffanteil in der Nahrung langsam erhöhen.
- Viel Obst und Gemüse essen sowie ein bis zwei Portionen fetten Seefisch pro Woche.
- Regelmäßig bewegen und mehr körperliche Aktivität in den Alltag einbauen. Fünfmal die Woche 30 Minuten mäßige Anstrengung wäre ein gutes Ziel.



Medikamente
Die erste Wahl bei der medikamentösen Therapie sind Statine. Sie blockieren die Cholesterin-Bildung in der Leber. Mit ihnen ist es gewöhnlich möglich, die LDL-Rezeptorfunktion der Leber zumindest teilweise wieder zu normalisieren, wodurch der LDL-Spiegel im Blut sinkt. Bei einer familiären Hypercholesterinämie mit entsprechenden Cholesterinwerten sollte die Behandlung mit Statinen bereits im Kindesalter beginnen.


Sollte die Therapie mit Statinen nicht ausreichend wirksam sein, wird häufig mit einem Cholesterin-Aufnahme-Hemmer mit dem Wirkstoff Ezetimib kombiniert. Dieses Medikament hemmt die Aufnahme von Cholesterin im Darm.
Die Bempedoinsäure ist ein Medikament, das die Cholesterinproduktion in der Leber hemmt. Nach bisherigen Studien lässt sich damit das LDL-Cholesterin um etwa ein Drittel senken. In Kombination mit Ezetimib sogar etwa um die Hälfte, sodass diese Kombination eine Behandlungsmöglichkeit für Patienten und Patientinnen dartstellt, die Statine nicht vertragen.
Wenn es trotz dieser verschiedenen Medikamente und ihrer Kombination innerhalb eines Jahres nicht gelingt, das LDL-Cholesterin ausreichend zu kontrollieren, gibt es bei einer bereits vorliegenden Herz-Kreislaufkrankheit oder einem hohen Risiko dafür die Möglichkeit, eine Behandlung mit sogenannten PCSK-9-Antikörpern oder Inclirisan zu beginnen. Beide Substanzen vermindern - wenn auch auf unterschiedliche Weise - den Abbau hepatischer LDL-Rezeptoren durch PCSK-9-Antikörper. PCSK-9-Antikörper werden alle zwei bis vier Wochen, Inclisiran nur halbjährlich unter die Haut gespritzt.

Grafik: Die Wirkung von PCSK9-Antikörpern.
© W&B/Dr. Ulrike Möhle
LDL-Apherese
Sollten Medikamente nicht genügen, um die LDL-Spiegel auf den erwünschten Wert zu senken, bleibt nur eine regelmäßige Blutwäsche, bei der LDL aus dem Blut entfernt wird (LDL-Apherese). Dabei wird Blut des Patienten über einen speziellen Schlauch in eine Maschine geleitet. Sie trennt unerwünschte Blutbestandteile ab – in diesem Fall das überschüssige LDL-Cholesterin. Das so "gereinigte" Blut erhält der Patient oder die Patientin wieder zurück. Die LDL-Apherese findet regelmäßig, beispielsweise alle zwei Wochen statt.

Unser Experte: Professor Dr. Wolfram Delius.
© W & B/Sabine Stallmann
Beratender Experte
Professor Dr. med. Wolfram Delius ist Facharzt für Innere Medizin und Kardiologie. Er habilitierte sich an der medizinischen Universitäsklinik Uppsala, Schweden, und hatte anschließend eine außerordentliche Professur für Medizin an der Technischen Universität München inne. Der Herzspezialist war lange Zeit als Chefarzt tätig, zuletzt zwei Jahrzehnte an der Abteilung Kardiologie/Pneumologie am Städtischen Krankenhaus München-Bogenhausen (Akademisches Lehrkrankenhaus). Inzwischen führt er eine eigene Praxis.
Profesor Delius wirkt seit Jahren aktiv bei Fortbildungsveranstaltungen der Bayerischen Ärztekammer mit und wurde mit der Ernst von Bergmann Plakette der Bundesärztekammer ausgezeichnet.
Wichtiger Hinweis: Dieser Artikel enthält nur allgemeine Hinweise und darf nicht zur Selbstdiagnose oder –behandlung verwendet werden. Er kann einen Arztbesuch nicht ersetzen. Die Beantwortung individueller Fragen durch unsere Experten ist leider nicht möglich.