

Mukoviszidose

Die Mukoviszidose (zystische Fibrose oder in anderer Schreibweise auch cystische Fibrose, häufig verwendete Abkürzung: CF) ist eine Erbkrankheit. Aufgrund eines veränderten Gens auf dem Chromosom Nummer 7 (CFTR-Gen) ist der Salz- und Wassertransport der Zellen gestört.
Die Sekrete vieler Körperdrüsen sind daher bei Mukoviszidose zähflüssiger als normal. Besonders schwerwiegend betroffen ist meist die Lunge. Der in den Bronchien gebildete Schleim lässt sich nur schwer abhusten, was die Belüftung der Lunge beeinträchtigt und gute Wachstumsbedingungen für Bakterien schafft. Die Folge: Menschen mit Mukoviszidose leiden meist nicht nur unter chronischem Husten mit eitrigem Auswurf sondern auch häufig unter Lungenentzündungen und anderen wiederkehrenden Atemwegsinfekten.
Mukoviszidose ist insgesamt eine seltene Erkrankung. Dennoch ist sie die häufigste tödlich verlaufende angeborene Stoffwechselkrankheit bei hellhäutigen Menschen in Europa und den USA.
Mukoviszidose ist nicht heilbar. Durch eine frühzeitige Therapie bereits im Kindesalter kann der Verlauf der Erkrankung jedoch positiv beeinflusst werden. Inzwischen liegt das mittlere Überleben bei über 40 Jahren. Ein heute geborenes Kind mit Mukoviszidose hat eine noch wesentlich bessere mittlere Lebenserwartung.
Was sind die Symptome einer Mukoviszidose?
Wie sich die Mukoviszidose im Einzelfall äußert, ist recht unterschiedlich. Betroffen sind in der frühen Kindheit vor allem die Bauchspeicheldrüse und die Verdauung, später stehen dann Atemwege, Lunge, Leber, Gallenwege und die Keimdrüsen im Vordergrund. Meist sind die Sekrete der Drüsen zäher als normal. Dies führt dazu, dass die Absonderungen nur schwer ausgeschieden oder abgehustet werden können oder gar den Ausführungsgang der Drüsen verstopfen.
Ein verspäteter erster Stuhlgang des Neugeborenen (Kindspech, Mekonium) oder gar ein Darmverschluss durch das Kindspech (Mekoniumileus) können erste Hinweise auf die Krankheit sein. Ein Mekoniumileus kann schon vor der Geburt auftreten.
Auch das Sekret der Bauchspeicheldrüse (Pankreas) ist zäher als normal und kann daher nicht entsprechend ausgeschieden werden. Die Funktion der Bauchspeicheldrüse ist damit oft eingeschränkt (exokrine Pankreasinsuffizienz). Das Pankreas bildet zahlreiche Stoffe, die bei der Verdauung helfen. Fehlen diese im Darm, weil nicht genügend Sekret ausgeschieden wird, können bestimmte Nahrungsbestandteile, wie zum Beispiel die Fette, nicht mehr richtig aufgenommen werden. Die Folge sind ein Nährstoffmangel sowie Durchfall. Der Stuhlgang ist oft sehr voluminös, fettig und übelriechend. Auf Dauer können sich Untergewicht und bei Kindern Wachstumsstörungen entwickeln. In manchen Fällen entsteht durch die Schädigung der Bauchspeicheldrüse zusätzlich eine Zuckerkrankheit (Diabetes mellitus).
Verdauungsbeschwerden sind bei Mukoviszidose nicht selten. Kinder, Jugendliche und Erwachsene kämpfen teilweise mit häufigen voluminösen Stuhlentleerungen oder auch mit Verstopfung. Ein Mastdarm-Vorfall (Rektum-Prolaps), bei dem der Enddarm aus dem After hervortritt, kommt bei Menschen mit cystischer Fibrose öfter vor als bei gesunden Personen.
Unbehandelt kommt es bereits im Säuglings- und Kindesalter zu Lungenentzündungen und wiederkehrenden Infekten der Atemwege wie chronische Bronchitis und Nasennebenhöhlenentzündungen. Die Ursache ist der zähe Schleim, der nur schlecht abgehustet werden kann und einen guten Nährboden für Keime bietet.
Aber auch in Leber und Gallenwegen macht sich die cystische Fibrose bemerkbar. Durch die erhöhte Zähigkeit der Gallenflüssigkeit kann es zum Gallenstau oder zu Gallesteinen kommen. Die Folge können Entzündungen der Leber und im weiteren Krankheitsverlauf auch Veränderungen der Struktur und damit der Funktionsfähigkeit des Organs in Form einer Leberzirrhose sein. Sie kommt etwa bei zehn Prozent der Erwachsenen mit Mukoviszidose vor.
Auch die Geschlechtsorgane sind von der Mukoviszidose betroffen. Bei Frauen mit cystischer Fibrose ist der Schleim im Gebärmutterhals zäher als bei Gesunden. Spermien können diesen zähen Schleim nur schwer durchdringen. Dies müssten sie aber, um die Eizelle im Eileiter befruchten zu können. Bei Männern werden durch die Mukoviszidose die Samenleiter in ihrer Entwicklung beeinträchtigt. Die Folge ist, dass zwar Spermien gebildet werden, ihnen aber der Weg aus dem Körper versperrt ist. Auf natürlichem Weg können daher viele Männer mit Mukoviszidose kein Kind zeugen. Eine künstliche Befruchtung ist jedoch zumeist möglich.
Ein Symptom der Mukoviszidose, das vor allem diagnostisch von Bedeutung ist, ist der erhöhte Salzgehalt im Schweiß, der sich mit bestimmten Tests nachweisen lässt.
Ursachen und Vererbung von Mukoviszidose
Mukoviszidose ist erblich. Die Grundlage der Krankheit ist eine Veränderung eines Gens auf dem Chromosom 7, des sogenannten CFTR-Gens. Die Vererbung einer Mukoviszidose erfolgt autosomal-rezessiv. Das bedeutet, dass nicht jeder, der ein verändertes CFTR-Gen in sich trägt, erkrankt. Im Normalfall leiden nur Personen an Mukoviszidose, die von beiden Eltern eine veränderte Version des Gens erhalten haben. Menschen, die nur ein verändertes Gen haben, können dieses zwar an ihre Kinder weitergeben, erkranken aber üblicherweise selbst nicht. Etwa jede 25ste Person besitzt ein verändertes CFTR-Gen. Mukoviszidose kommt in Europa mit einer Häufigkeit von etwa 1: 2.500 vor. Obwohl beim CFTR-Gen weit mehr als 2000 verschiedene Mutationen bekannt sind, ist bei mehr als 70 Prozent der Betroffenen in Westeuropa eine bestimmte Veränderung vorhanden: Die Mutation F508del, auch mit dem Rufnamen Delta-F-508 bezeichnet.
Gene leiten den Körper zum Bau bestimmter Eiweiße an. Das CFTR-Gen steht für einen Kanal in der Zellwand, durch den Chlorid-Ionen transportiert werden. Bei Mukoviszidose ist dieser Kanal defekt oder nicht vorhanden. Die Folge sind Veränderungen der Salz- und Wasserströme in und aus den Zellen. Diese führen dazu, dass verschiedene Drüsensekrete zähflüssiger sind als normal und dass der Salzgehalt im Schweiß erhöht ist.
Diagnose
Ein Schweißtest war früher die einzige Möglichkeit, eine Mukoviszidose zu diagnostizieren. Denn der Salzgehalt im Schweiß ist aufgrund des defekten Chloridkanals bei Menschen mit cystischer Fibrose erhöht. Findet sich im sogenannten Pilocarpin-Iontophorese-Schweißtest eine erhöhte Salzkonzentration, so ist es sehr wahrscheinlich, dass tatsächlich eine zystische Fibrose vorliegt.
Sind die Ergebnisse des Schweißtests nicht eindeutig und besteht trotzdem der Verdacht auf eine Mukoviszidose, kann die Messung einer Potentialdifferenz eventuell weiterhelfen. Denn durch die Veränderungen im Salz- und Wasserhaushalt ergeben sich bei bestimmten Tests messbare elektrische Unterschiede im Vergleich zum Gesunden. Eine Messung von Potentialdifferenzen ist an der Nasenschleimhaut oder aber an einer Gewebeprobe (Biopsie) aus dem Mastdarm (Rektum) möglich.
Eine genetische Untersuchung mit Nachweis der Veränderung im CFTR-Gene liefert nicht nur Gewissheit, sondern kann auch genau zeigen, um was für eine Veränderung des Gens es sich handelt. Für den Gentest ist nur eine kleine Menge Blut notwendig.
Zusätzlich ist es wichtig, einen Überblick darüber zu erhalten, welche Organe bereits wie schwer von der Krankheit betroffen sind. Dazu kommen verschiedene Untersuchungen, wie zum Beispiel Lungenfunktionstests, bildgebende Verfahren wie Röntgen-, MRT- oder Ultraschalluntersuchungen oder Untersuchungen der Funktion der Bauchspeicheldrüse anhand von Blut- und Stuhltests in Frage. Eine Analyse der Absonderungen aus den Atemwegen (Auswurf/Sputum) hilft zu erkennen, ob die Lunge mit krankmachenden Keimen infiziert ist. Hier steht besonders das Bakterium Pseudomonas aeruginosa im Fokus. Aber auch viele andere Erreger, die bei Menschen mit zystischer Fibrose Atemwegsinfektionen hervorrufen können, sind bedeutsam. Im weiteren Verlauf finden dann immer wieder entsprechende Kontrolluntersuchungen statt.
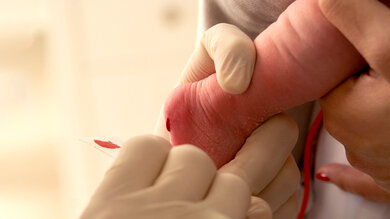
Mit dem beim Neugeborenen-Screening gewonnen Blut wird auch ein Test auf Mukoviszidose gemacht
© iStockphoto/Odairson Antonello
Früherkennung
In Deutschland wird seit 2016 für alle Neugeborenen eine Untersuchung auf Mukoviszidose angeboten, um die Krankheit möglichst früh zu diagnostizieren. Diese Untersuchung findet üblicherweise zusammen mit dem Neugeborenen-Screening und aus derselben Blutprobe statt.
Im Labor wird aus dem auf Filterpapier getropften Blut zunächst das sogenannte immunreaktive Trypsin (IRT) bestimmt. Ist es sehr hoch, so wird ein Schweißtest empfohlen. Bei einem nicht eindeutigen IRT-Wert wird aus derselben Blutprobe auch noch das Pankreatitis-assoziierte Protein (PAP) bestimmt. Ist auch dieser Wert erhöht, wird als nächstes in derselben Probe nach den Erbgutveränderungen gesucht, die häufig bei Mukoviszidose auftreten. Wird mindestens eine entsprechende krankhafte Veränderung gefunden, gilt das Ergebnis als positiv. Dies bedeutet aber keine Diagnose, sondern lediglich die Notwendigkeit einer weiteren Abklärung.
Die Eltern betroffener Kinder werden dann an spezialisierte Mukoviszidose-Zentren überwiesen, wo dann ein Schweißtest erfolgt. Ein kontrollbedürftiges Ergebnis bedeutet also noch nicht, dass das Baby tatsächlich an zystischer Fibrose leidet. Das ist nur bei circa einem von fünf Kindern mit kontrollbedürftigem Ergebnis der Fall.
Grundsätzlich besteht auch die Möglichkeit, das Ungeborene vor der Geburt (pränatal) auf das Vorhandensein genetischer Störungen und so auch des veränderten CFTR-Gens zu testen. Dazu wird zu einem bestimmten Zeitpunkt in der Schwangerschaft Fruchtwasser (Amniozentese) oder ein Stück des Mutterkuchens (Chorionzottenbiopsie) entnommen und darin das entsprechende Gen untersucht. Da dieses Vorgehen mit Risiken für das ungeborene Kind behaftet ist, sollten sich Frauen, die eine solche Untersuchung in Erwägung ziehen, zuvor ausführlich beraten lassen.
Therapie

Inhalieren: Wiederkehrende Atemwegsinfekte sind nur eines von vielen Symptomen der zystischen Fibrose.
© dalibor - stock.adobe.com
Die Behandlung richtet sich sehr danach, welche Organe betroffen sind und welche Symptome vorliegen. Was für die eine Person sinnvoll ist, ist für die andere nicht zwingend ebenfalls geeignet. Da Mukoviszidose eine seltene Erkrankung ist, empfiehlt es sich, spezielle Mukoviszidosezentren mit entsprechender Erfahrung aufzusuchen.
Da die Aufnahme von Nährstoffen aus dem Verdauungstrakt bei Mukoviszidose gestört sein kann, ist eine ausgewogene, relativ kalorienreiche Ernährung in Verbindung mit der Zufuhr von Verdauungsenzymen besonders wichtig, um Untergewicht und Mangelerscheinungen vorzubeugen. Eventuell kann unterstützend auch eine spezielle Kost oder die Verabreichung bestimmter Nährstoffe notwendig sein. Wichtig ist es auch, dem Körper ausreichend Salz zuzuführen. Betroffene sollten sich dazu und zu allen weiteren Therapiemöglichkeiten am besten bei ihrem behandelnden Arzt oder der Ärztin informieren! Er oder sie kann eventuell auch entsprechend spezialisierte Ansprechpartner und -partnerinnen vermitteln (zum Beispiel Ernährungsberater und -beraterinnen oder Physiotherapeutinnen und Physiotherapeuten).
Unter anderem kommen folgende Therapieansätze in Betracht:
- Atemgymnastik und Atemtherapie: Damit kann die Lungenfunktion verbessert und Infekten vorgebeugt werden.
- Schleimlösende Verfahren: Hier kommen verschiedene Möglichkeiten in Frage. Sie werden oft auch kombiniert eingesetzt. Autogene Drainage (ein Verfahren, bei dem der Patient lernt, ohne zu husten das Sekret gezielt und wirksam aus der Lunge hervorzubringen), reflektorische Atemtherapie und andere krankengymnastische Maßnahmen helfen, den zähen Schleim zu lockern und aus der Lunge zu entfernen. Inhalationen – zum Beispiel mit bestimmten schleimlösenden Mitteln – können ebenfalls sinnvoll sein. Ein weiterer Ansatz ist die Inhalation mit Kochsalzlösung oder DNAse, einem Enzym das die Zähflüssigkeit des Schleims durch die Spaltung der enthaltenen Bestandteile reduziert.
- Atemwegsinfekte müssen gezielt und konsequent behandelt werden. Dazu können häufige, teilweise auch regelmäßig wiederkehrende Behandlungen mit Antibiotika nötig sein. Pseudomonas aeruginosa ist ein Bakterium, das Gesunden relativ wenig anhaben kann, aber bei Patienten mit cystischer Fibrose oft zu Infektionen führt. Deswegen ist es bedeutsam, eine mögliche Infektion mit diesem Keim festzustellen. Eine Inhalation mit bestimmten Antibiotika kann dann zum Beispiel einer dauerhaften Besiedlung der Atemwege mit diesem Keim entgegengewirken.
- Bei Verengungen der Bronchien können eventuell bronchienerweiternde Medikamente hilfreich sein.
- Kann die Bauchspeicheldrüse nicht mehr die für eine gesunde Verdauung notwendigen Stoffe bilden, müssen diese in Tablettenform eingenommen werden. Eine solche Enzymersatztherapie hilft, Mangelernährung und Untergewicht vorzubeugen. Da insbesondere die Aufnahme der fettlöslichen Vitamine A, D, E und K aus dem Darm gestört sein kann, müssen diese eventuell zusätzlich verabreicht werden.
- Verstopfungen lassen sich durch Einläufe oder Abführmittel in den Griff bekommen.
- Bei einer Leberzirrhose durch Gallestau kann eine Behandlung mit dem Wirkstoff Ursodesoxycholsäure sinnvoll sein.
- Nehmen Luftnot und Atembeschwerden zu und die Lungenfunktion stetig ab, kann eine Therapie mit Sauerstoff notwendig sein.
Die Forschung gewinnt einen immer genaueren Überblick, welche Folgen bestimmte Gendefekte, die eine Mukoviszidose hervorrufen, im Einzelnen für den Körper haben. Das macht die Entwicklung von Therapien möglich, die diese Störungen umgehen. Erste CFTR-Medikamente, sogenannte Modulatoren, wurden bereits identifiziert und zur Therapie zugelassen. Sie verändern keineswegs das Erbgut der Patienten, aber sie korrigieren bestimmte Defekte des CFTR-Kanals, etwa eine fehlgefaltete Eiweisstruktur, so dass diese wieder funktioniert.
Ist zum Beispiel das Eiweiss so gefaltet, dass es an der Zelloberfläche seiner Funktion als Salzpumpe nicht nachkommt, dann können Potentiatoren diese Funktion wiederherstellen. Der erste Potentiator, Ivacaftor, wurde 2012 für die Mutation G551D zugelassen. Durch Ivacaftor wird bei den von dieser Mutation betroffenen Menschen eine deutliche Linderung der Symptome erreicht.
Für die häufigste genetische Veränderung bei Menschen mit Mukoviszidose (F508del) gibt es für Kinder ab zwei Jahren zugelassene Kombinationen, sogenannte Korrektoren und Potentiatoren, die die defekten Eiweiße an Zellkontrollpunkten vorbeischleusen und so den Einbau in die Zellmembran ermöglichen.
Der Einsatz dieser neuen Kombinationen aus Korrektoren (Lumacaftor, Elexacaftor, Tezacaftor) und Potentiatoren (Ivacaftor) verbessert die Lebenserwartung der Patienten deutlich. Obwohl damit erstmals eine ursächliche Behandlung der Mukoviszidose möglich ist, ist die Erkrankung aber weiterhin nicht heilbar und erfordert eine lebenslange intensive Therapie. Darüber hinaus gibt es Mutationen, die man mit den Modulatoren nicht behandeln kann. Menschen, die eine solche Mutationskombination haben, sind weiterhin nicht ursächlich behandelbar. Man schätzt, dass das in Deutschland etwa 10 Prozent der Menschen mit Mukoviszidose sind.
Bei schwerer Beeinträchtigung der Lungenleistung kommt einen Lungentransplantation in Betracht. Zwar treten an einer transplantierten Lunge nach einer Lungentransplantation keine Mukoviszidose-Probleme auf, da diese ja die gesunden Gene des Spenders besitzt. Allerdings ist diese Maßnahme auch keine Heilung und die Nachsorge und Therapie für den Patienten mit einer neuen Lunge sehr aufwändig.
Ein erfolgreicher Einsatz einer Gen-Therapie, bei der das kranke CFTR-Gen gegen ein gesundes ausgetauscht wird, ist noch in der Erprobungsphase. Dieses Verfahren verspricht Hoffnung für die Zukunft, auch wenn man heute noch nicht abschätzen kann, wann es wirklich zur Behandlung von Patienten eingesetzt werden kann.
Beratender Experte
Dr. med. habil. Olaf Eickmeier ist Facharzt für Kinder- und Jugendmedizin und Kinderpneumologe. Er leitet die pädiatrische Sektion des Christiane Herzog CF-Zentrums in Frankfurt am Main und ist Oberarzt am Universitätsklinikum Frankfurt am Main. Er studierte Medizin an der Rheinischen Friedrichs-Willhelms-Universität Bonn, wo er 2004 auch promovierte. Thema seiner Doktorarbeit war die anti-entzündliche Wirkung von Montelukast auf milde Verlaufsformen der zystischen Fibrose. 2021 habilitierte er sich an der Johann Wolfgang von Goethe-Universität in Frankfurt am Main.

Unser Experte: Dr. med. habil. Olaf Eickmeier
© privat


