

Mit der Genschere ans Blut

Bei der Sichelzellkrankheit sind Teile des Hämoglobins defekt - die Genschere Crispr-Cas kann hier helfen.
© mauritius images / Science Photo
Warum wird jetzt darüber geredet?
Erbkrankheiten mit einem präzisen Schnitt im Erbgut korrigieren: So lautet seit zehn Jahren das medizinische Versprechen der Genschere Crispr-Cas. Seit April laufen nun die Zulassungsverfahren für eine Gentherapie der Sichelzellkrankheit. Ursache der Erkrankung ist eine Punktmutation. Sie macht den roten Blutfarbstoff Hämoglobin in den roten Blutkörperchen, den Erythrozyten, schwer löslich. Der Farbstoff bildet Fasern und bindet nur noch wenig Sauerstoff, so dass die Blutkörperchen sichelförmig werden. Sie gehen leicht kaputt oder können sich ineinander verhaken und Gefäße verschließen.
Das führt zu Infarkten in Geweben und Organen, den Sichelzellkrisen. Anstrengungen oder dünne Luft befördern diese Krisen. Bereits bei Kindern können Schlaganfälle auftreten. Bluttransfusionen und Medikamente wirken dem entgegen, sind aber lebenslang notwendig.
„Der bisher einzige heilende Ansatz ist die Knochenmarktransplantation“, sagt Dr. Lena Oevermann. Sie leitet an der Berliner Charité die größte deutsche Ambulanz für Erkrankungen, die mit dem roten Blutfarbstoff zusammenhängen. Die meisten Betroffenen finden Oevermann zufolge keinen passenden Spender. Und wer eine Spende erhält, muss Risiken in Kauf nehmen. „Unter anderem können Immunzellen des Transplantats den Körper des Behandelten angreifen“, so die Expertin. Zuvor schafft eine Chemotherapie Platz im Knochenmark. Sie kann zu Unfruchtbarkeit führen, insbesondere wenn Transplantation und Chemo wiederholt werden müssen.
So funktioniert die Genschere
© W&B/Dr. Ulrike Möhle
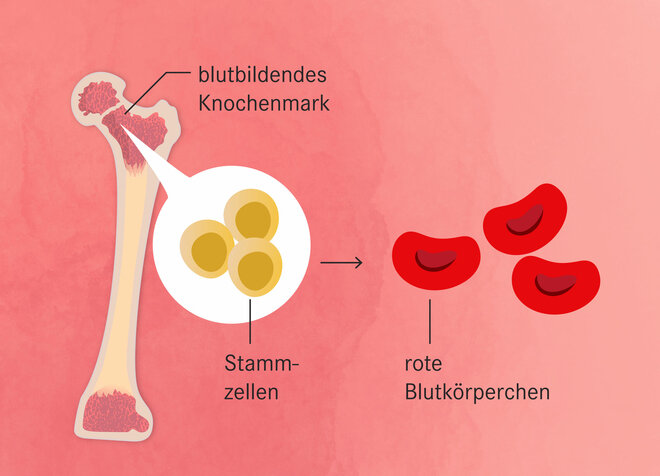
Die Produktion von Hämoglobin
Rote Blutkörperchen (Erythrozyten) werden im Knochenmark gebildet.
Sie produzieren große Mengen Hämoglobin, den roten Blutfarbstoff, aus dem Erythrozyten zu 90 Prozent bestehen.
© W&B/Dr. Ulrike Möhle
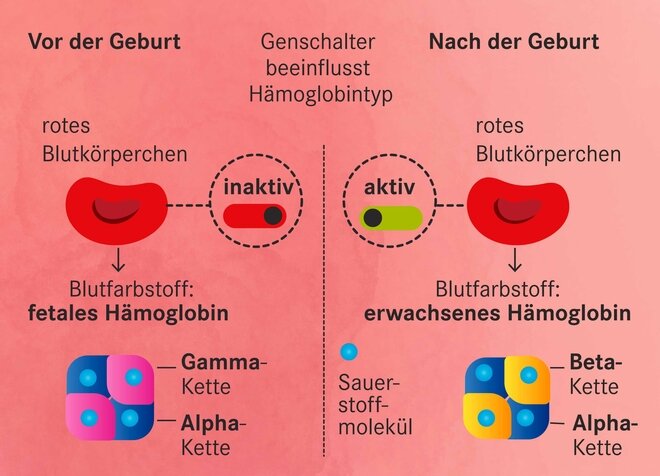
Der Farbstoff im Wechsel
Bei Gesunden verändert der rote Blutfarbstoff nach der Geburt die Zusammensetzung.
Beim Ungeborenen besteht das Hämoglobin aus je zwei Eiweißketten des Typs Alpha und des Typs Gamma.
Nach der Geburt wird ein genetischer Schalter umgelegt – statt Gamma-Ketten enthält das Hämoglobin nun Beta-Ketten.
© W&B/Dr. Ulrike Möhle
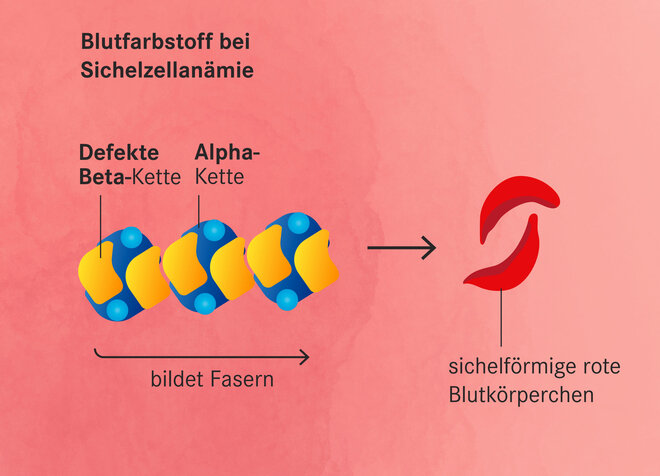
Ein fehlerhafter Farbstoff
Bei der Sichelzellkrankheit sind Teile des Hämoglobins defekt.
Eine Veränderung im Erbgut erzeugt klebrige Beta-Ketten, Das Hämoglobin bildet nun Fasern. Es nimmt weniger Sauerstoff auf und verändert die Form der roten Blutkörperchen.
© W&B/Dr. Ulrike Möhle
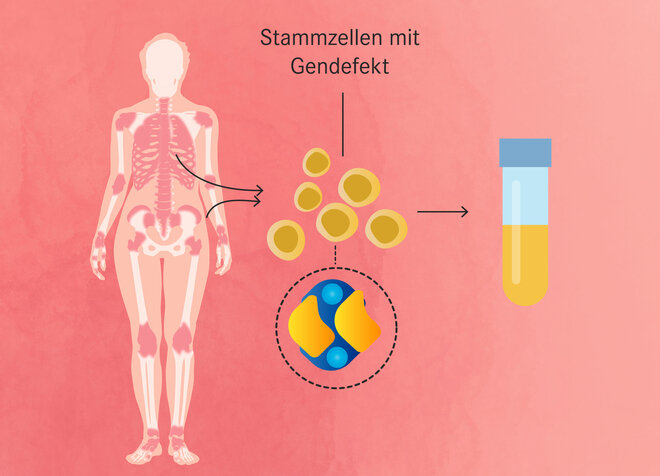
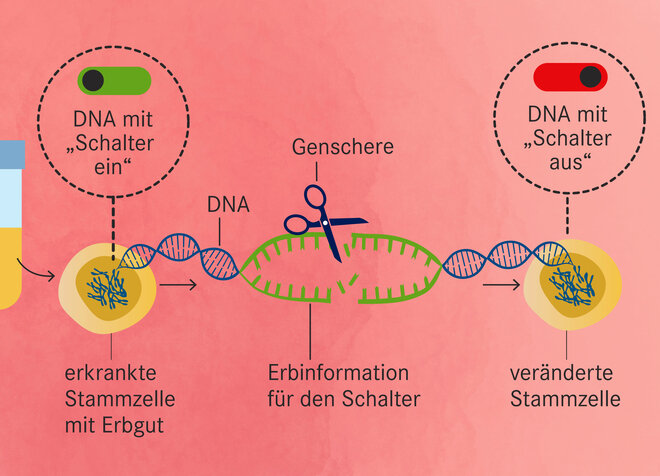
Der Trick...
Die Gentherapie setzt am Knochenmark an.
Im ersten Schritt werden die Blutstammzellen aus dem Knochenmark ins Blut geschwemmt und herausgefiltert.
© W&B/Dr. Ulrike Möhle
... mit der Schere
Die Genschere Crispr-Cas kann den Gendefekt zwar nicht reparieren. Aber sie kann das fetale Hämoglobin reaktivieren.
Der Schalter, der das fetale Hämoglobin unterdrückt, wird wie das Hämoglobin in der DNA codiert.
Die Genschere besitzt eine Schablone, mit der sie am Gen für den Schalter ansetzt. Sie schneidet. Der Schalter geht aus.
Die therapierten Stammzellen können jetzt fetales, funktionales Hämoglobin produzieren, wenn sie rote Blutkörperchen bilden.
© W&B/Dr. Ulrike Möhle
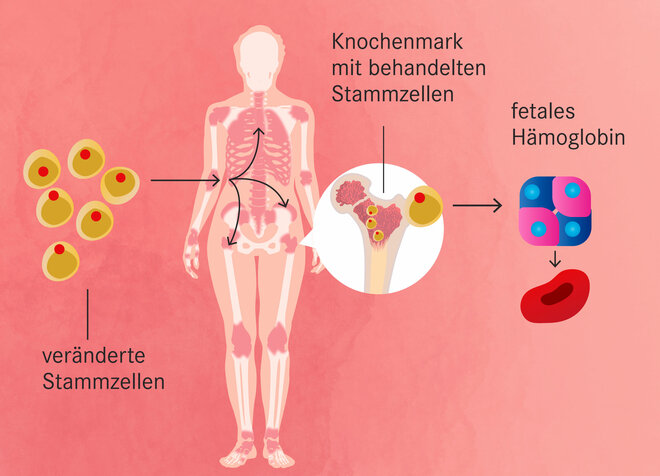
Der fetale Effekt
Die Stammzellen wandern zurück ins Knochenmark. Sie bilden normal geformte rote Blutkörperchen mit fetalem Hämoglobin.
Was ist das Besondere daran?
„Man würde meinen, dass die Genschere Crispr-Cas die Punktmutation korrigieren kann, die für die Sichelzellkrankheit verantwortlich ist“, sagt Lena Oevermann. Tatsächlich ist das bisher nur im Tierversuch gelungen. Im Menschen richtet sich die Genschere daher gegen einen Schalter, der nach der Geburt die Produktion von fetalem Hämoglobin abstellt. Es bindet Sauerstoff besonders gut, nach der Geburt wird es von Gesunden nicht mehr benötigt. Einige Menschen mit Sichelzellanämie profitieren aber davon, dass ihre Blutzellen aufgrund zusätzlicher Erbveränderungen weiter fetalen Blutfarbstoff produzieren. Er enthält kein fehlerhaftes Eiweiß. Die Betroffenen sind fast symptomfrei, obwohl der Erbdefekt im Erwachsenen-Hämoglobin noch da ist.
Genau diese Kombination stellt die neue Gentherapie nach. Sie korrigiert zwar den Erbdefekt nicht. Aber sie schaltet durch einen Schnitt der Genschere die Produktion von fetalem Hämoglobin an. Dafür werden Stammzellen aus dem Knochenmark der Erkrankten entnommen, mit der Genschere behandelt und zurückgegeben. Studien zufolge erleben neun von zehn Menschen mit Sichelzellanämie nach der Behandlung keine Sichelzellkrisen mehr. „Es ist keine Heilung, aber eine funktionelle Korrektur“, sagt Oevermann. Das Risiko der Unfruchtbarkeit allerdings bleibt: Auch vor der Rückkehr der behandelten Stammzellen muss das verbliebene Knochenmark durch eine Chemotherapie beseitigt werden.
Welche Kranken profitieren davon?
Nur wenige Erkrankte werden die neue Therapie erhalten. Die Kosten je Behandlung sollen bei mehr als einer Million Dollar liegen. Für westliche Staaten ist das viel – für Länder in Afrika, wo viele Menschen die Sichelzellkrankheit haben, ist das unerschwinglich. „Die Menschen dort bekommen nicht einmal die Standardversorgung“, sagt Oevermann, die in Kenia beratend tätig ist. In Deutschland gebe es neuen Schätzungen zufolge etwa 6000 Menschen mit Sichelzellkrankheit. „Weltweit aber werden jährlich 350 000 Babys mit dieser Krankheit geboren.“ Sie wünsche sich, dass auch diese Kinder die bestmögliche Behandlung erhalten.


Quellen:
- Page ML: Sickle cell disease is now curable, but the treatment is una!ordable. New Scientist: https://www.newscientist.com/... (Abgerufen am 26.06.2023)
- Oevermann L, Sodari P: Status quo of allogeneic stem cell transplantation for patients with sickle cell disease using matched unrelated donors. In: Hematol. Oncol Stem Cell Ther. 13.06.2020, 13: 116-119
- Frangoul H et al.: CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. In: NEJM 21.01.2021, 384: 252-260
- Byrne J: CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. BioPharmaReporter: https://www.biopharma-reporter.com/... (Abgerufen am 26.06.2023)
-
Pressemitteilung Crispr Therapeutics