

Myasthenie

Was ist eine Myasthenie?
Bei einer Myasthenie handelt es sich um eine Erkrankung, die durch eine fehlerhafte Signalübertragung der Nerven auf die Muskulatur charakterisiert ist. Sie fällt durch eine belastungsabhängige Muskelschwäche auf, die sich in Ruhe wieder bessert. Meist kommt es zu einer Zunahme der Beschwerden in der zweiten Tageshälfte. Durch Infektionen, extreme Hitze oder Kälte, bestimmte Medikamente, Stress oder kurz vor der Periode können die Beschwerden verstärkt auftreten.
Neben der autoimmun-vermittelten Myasthenia gravis (MG) kommen zudem genetisch bedingte und somit angeborene Muskelschwächen (kongenitale myasthene Syndrome = CMS) vor. In den vergangenen Jahrzehnten ist die Inzidenz der MG gestiegen, was allerdings auch durch eine bessere Diagnostik erklärt werden kann. Der Häufigkeitsgipfel liegt zwischen dem 60. und 80. Lebensjahr. Circa 10 Prozent der Betroffenen sind Kinder und Jugendliche unter 17 Jahren (juvenile MG). Frauen sind bei der früh-einsetzenden (early-onset) MG häufiger betroffen als Männer (Verhältnis 3:2). Bei der spät-einsetzenden (late-onset) MG sind Männer etwas häufiger betroffen. Liegt eine Erkrankung des Thymus zugrunde (siehe Ursachen), ist das Geschlechterverhältnis gleich und die Krankheit tritt vor allem zwischen dem 50. und 70. Lebensjahr auf.
Meist kommt es zu einer Zunahme der Beschwerden in der zweiten Tageshälfte. Durch Infektionen, extreme Hitze oder Kälte, bestimmte Medikamente, Stress oder kurz vor der Periode können die Beschwerden verstärkt auftreten.
Neuromuskuläre Signalübertragung
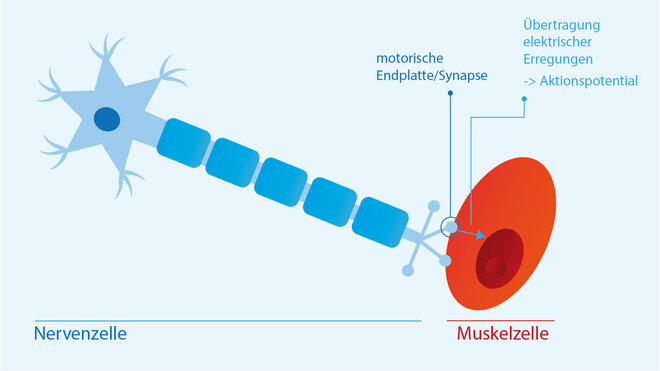
© W&B/Christina Angele
Das Bild zeigt die Übertragung eines Nervensignals auf einen Muskel. Nervensignale (Aktionspotentiale) werden über Veränderung von Spannungszuständen (Polaritäten) aufgrund Ionenverschiebungen weitergeleitet. Am Ende des Nerven befindet sich eine Synapse (Endknöpfchen der Nervenzelle), welche in einen sehr geringen Abstand zur Muskulatur endet.
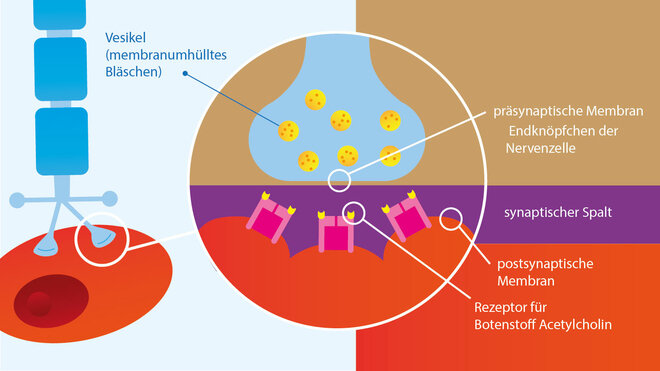
© W&B/Christina Angele
Dieser sehr geringe Abstand ist der synaptische Spalt. Damit nun die elektrische Übertragung weiterläuft, wird hierfür ein Botenstoff benötigt. Dieser Botenstoff ist das Acetylcholin (ACh), welcher an der Nervenendigung in membranumhüllten Bläschen (Vesikel) gespeichert wird.
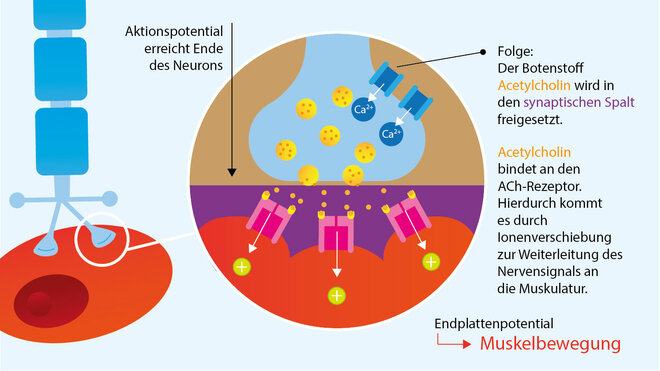
© W&B/Christina Angele
Nach Eintreffen eines Aktionspotentials wird nun durch Öffnung von Calcium-Kanälen die Spannung verändert und der Botenstoff Acetylcholin (ACh) aus den Vesikeln in den synaptischen Spalt freigesetzt. Auf der Muskelseite bindet nun ACh an den ACh-Rezeptor. Hierdurch kommt es erneut zu Ionenverschiebungen und damit zur Weiterleitung des Signals an die Muskulatur. Die Reaktion ist eine Muskelkontraktion.
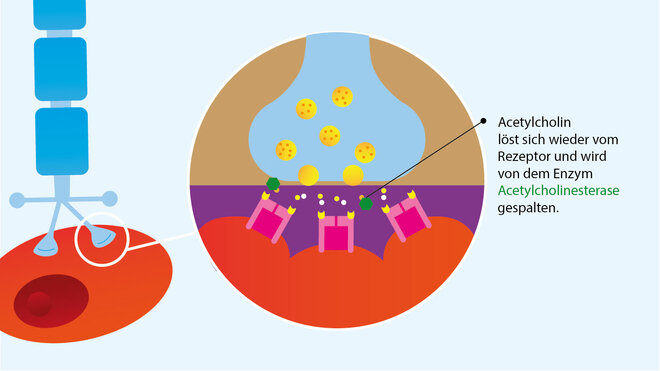
© W&B/Christina Angele
Nach einer kurzen Zeit löst sich ACh wieder vom Rezeptor und wird im synaptischen Spalt von dem Enzym Acetylcholinesterase in seine beiden Bestandteile Acetat und Cholin gespalten.
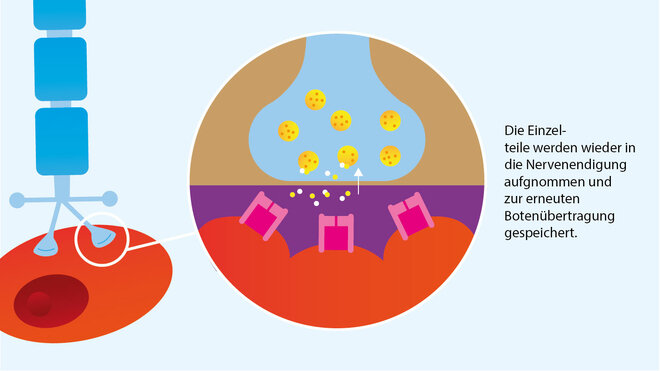
© W&B/Christina Angele
Die Bestandteile werden wieder in das Endknöpfen aufgenommen, zusammengesetzt und wieder in den Bläschen (Vesikel) gespeichert. Somit stehen sie für eine weitere Aktion zur Verfügung.
Ursachen: Wodurch kommt es zur Myasthenie?
Verschiedene Krankheiten führen zu einer Störung der neuromuskulären Erregungsübertragung (siehe oben). Am häufigsten ist die Myasthenia gravis, deutlich seltener ist das Lambert Eaton-Syndrom. Äußerst selten kommen verschiedene angeborene (kongenitale) myasthene Syndrome vor.
- Myasthenia gravis
Bei der Myasthenia gravis bildet der Körper meist Antikörper gegen körpereigene Bestandteile (Autoantikörper). Sie richten sich am häufigsten gegen den AChR (siehe Bildergalerie). Normalerweise dockt an diesen Rezeptor das benötigte ACh an, wie bei einem Schlüssel-Schloss-Prinzip.

Die produzierten Autoantikörper passen aber ebenso an den Rezeptor, besetzen also quasi ein Schlüsselloch, führen aber nicht zu einer Übermittlung des Nervensignals, öffnen also keine Tür. Somit stehen dem ACh weniger Rezeptoren zur Verfügung, an die sie andocken können. Neben dieser direkten Konkurrenz führen noch weitere Mechanismen zu einer Verminderung der Anzahl von Rezeptoren. Zum einen kommt es über die Aufnahme des Rezeptors in die Muskelzelle und den Abbau des Rezeptors (Endozytose) zu einem zusätzlichen Verlust von Andockstellen. Zum anderen greifen bestimmte Eiweiße (Bestandteile des Komplementsystems) die Zellmembran an der Muskelzelle an und schädigen sie, was dazu beiträgt, dass die Zahl der der ACh-Rezeptoren (AChR) abnimmt und eine Verbreiterung des synaptischen Spaltes bedingt. Die Folge: ACh muss länger durch den nun breiteren Spalt „wandern“, um zur motorischen Seite zu gelangen und wird bereits abgebaut, bevor es den Rezeptor erreicht.
Es stehen also aus mehreren Gründen weniger funktionstüchtige ACh-Rezeptoren zur Verfügung. In Summe bedeutet das: Weniger ACh erreicht sein Ziel und kann Signale übermitteln. Insbesondere bei sich wiederholenden Bewegungen führt nicht jeder Nervenimpuls zu einer Muskelbewegung, da der Schwellenwert zur Weiterleitung des Signals nicht erreicht wird. Dieser zeitweilige Ausfall einzelner Muskelfasern führt zu einer verminderten Kraft des gesamten Muskels.
Neben dem Autoantikörper gegen den AChR konnten auch weitere ursächliche Antikörper gefunden werden, wie MuSK oder LRP4. Ob und welche Antikörper bei MG-Patienten vorhanden sind, wird über Blutuntersuchungen geklärt.
Ursachen für die Autoantikörper bei MG
Die Erkrankung entsteht oft bei Menschen mit einer gewissen Veranlagung. Eine zentrale Rolle für die Ausbildung der Antikörper spielt der Thymus. Er sitzt hinter dem oberen Teil des Brustbeins und gehört zu den Organen des Immunsystems. Erkrankungen des Thymus wie Entzündungen (Thymitis) oder Tumore (Thymome) sollten deshalb ausgeschlossen beziehungsweise abgeklärt werden. Dazu soll jeder Patient mit der Diagnose Myasthenia gravis eine Untersuchung des Thymus mittels bildgebenden Verfahren (CT- oder MRT-Untersuchung) erhalten. Bei 15 Prozent der Betroffenen sind bösartige Tumoren des Thymus nachweisbar.
- Lambert Eaton-Syndrom
Beim Lambert-Eaton-Syndrom (Lambert Eaton Myasthenie-Syndrom = LEMS) handelt es sich ebenfalls um eine Autoimmunerkrankung, bei welcher sich Autoantikörper gegen die Calcium-Kanäle der präsynaptischen Membran richten (Lage siehe Bildergalerie). Aufgrund der verminderten Funktion dieser Kanäle kann die Übertragungskette nicht mehr richtig funktionieren, es kommt zu einer verminderten Ausschüttung von ACh, wodurch weniger dieses Botenstoffes zur Verfügung steht. Dadurch sind die muskuläre Erregung und Kontraktion erschwert.
Ursachen für die Autoantikörper bei Lambert-Eaton-Syndrom
Das LEMS kann tumor-assoziiert (paraneoplastisch) oder im Zusammenhang mit einer zugrundeliegenden Autoimmunerkrankung (idiopathisch, iLEMS) auftreten. Die paraneoplastische Form findet sich bei weniger als 50% der Betroffenen. In diesen Fällen ist zumeist eine Krebserkrankung der Lunge (kleinzelliges Bronchialkarzinom, SCLC) für die Bildung der Autoantikörper verantwortlich. Der Körper bildet eigentlich Antikörper gegen die Calcium-Kanäle des Tumors. Diese richten sich aber auch gegen die Calcium-Kanäle der präsynaptischen Membran.
- kongenitale myasthene Syndrome (CMS)
Hierbei handelt es sich um ein sehr uneinheitliches Krankheitsbild. Allen Formen gemeinsam ist, die neuromuskuläre Signalübertragung nicht immunvermittelt, sondern aufgrund genetischer Defekte beeinträchtigt ist. Dies bedingt Veränderungen allgegenwärtiger Proteine (Eiweißstoffe), welche an der neuromuskulären Übertragung beteiligt sind. Allerdings gibt es nicht „das eine Protein“, das CMS verursacht, sondern es ist individuell sehr unterschiedlich, welches Protein von der Veränderung betroffen ist.
Symptome: Welche Beschwerden bereitet eine Myasthenie?
Auffällig ist eine Ermüdung der Muskulatur, die im Laufe des Tages zunimmt. Bei der Myasthenia gravis sind vor allem kleinere Muskelgruppen betroffen, wie beispielsweise die Augenmuskulatur. Den Betroffenen fällt es schwer, die Augenlider zu öffnen oder es entstehen Doppelbilder nach längerem Lesen. Auch ein zunehmend herabhängendes Augenlid (auf einer Seite ausgeprägter als auf der anderen) kann auffallen. Diese Form der Myasthenie wird okuläre Myasthenie genannt, da sie zunächst die Augenmuskulatur betrifft (okulär = das Auge betreffend). Bei ca. 70-80 Prozent breitet sich die Schwäche meist innerhalb von zwei bis drei Jahren auch auf andere Muskelgruppen aus, man spricht von der Generalisierung der MG.
Bei der generalisierten Myasthenie können potenziell alle Muskeln beteiligt sein, typischerweise sind aber die stammnahen Muskeln von Armen und Beinen betroffen. Erkrankte haben daher zum Beispiel Schwierigkeiten bei Überkopfarbeiten wie dem Wäsche-Aufhängen oder dabei, Dinge über Kopf einzuräumen. Auch das Aufstehen aus einem Sessel oder Treppensteigen kann Probleme bereiten.
Allerdings können auch weitere Muskelgruppen beeinträchtigt sein, wie beispielsweise die Muskulatur des Gesichts, wodurch die Ausdrucksfähigkeit und Mimik abnimmt und „einfriert“. Ungünstiger wird es, wenn die Muskeln von Zunge, Schlund oder Kehlkopf betroffen sind (bulbäre Muskulatur). Klinische Zeichen für diese Form einer Myasthenie sind:
- Schwierigkeiten beim Kauen von harten oder zähen Speisen
- häufiges Verschlucken oder
- eine verwaschene Sprache.
Auch die Atemmuskulatur kann gestört sein. Das kann folgende Anzeichen haben:
- schwache, tonlose Stimme
- schwacher Hustenstoß oder
- häufiges Zwischenatmen beim Sprechen.
Bei dieser Erkrankungsform besteht ein erhöhtes Risiko für eine myasthene Krise: Sie gefährdet Betroffene durch eine Atemlähmung und schwere Schluckstörungen. Auslöser einer myasthenen Krise können eine unzureichende Behandlung, akute Infekte oder bestimmte Medikamente sein (siehe hierzu auch den Abschnitt „Was müssen Patienten mit Myasthenie beachten“).
Beim Lambert-Eaton-Syndrom ähneln die Symptome denen der Myasthenia gravis. Die Augenmuskeln sowie die Schlund- und Atem-Muskulatur sind allerdings nur selten betroffen, dafür häufiger die stammnahe Beinmuskulatur.
Auch das kongenitale myasthene Syndrom zeichnet sich durch Muskelschwäche aus. Es kann sich durch ein herabhängendes Augenlid, Schielen, eine abgeflachte Mimik oder einer Verzögerung der motorischen Entwicklung bemerkbar machen. Auch eine Trinkschwäche oder Störungen der Atemmuskulatur können auffallen.
Diagnose: Wie wird eine Myasthenie festgestellt?
- Anamnese und körperliche Untersuchung
Bereits die Krankengeschichte (Anamnese) mit charakteristischen Beschwerden lenkt den Verdacht auf eine Myasthenie. Zur näheren Differenzierung können spezielle Tests, welche zur Ermüdung der Muskulatur führen, herangezogen werden.
- Neurophysiologische Testverfahren
Neurophysiologische Tests zur Messung und Bestimmung der Muskelerregung (Elektromyografie, EMG) ergänzen die Diagnostik.
- Laboruntersuchungen
Mithilfe spezieller Laboranalysen des Blutes wird nach Antikörpern gesucht. So werden unter anderem Antikörper gegen den AChR, die muskelspezifische Kinase (MuSK) oder die Rezeptoren für spezifische Proteine (Lipoprotein-related protein 4, LRP-4) bestimmt, um die Myastenia gravis zu bestätigen.
Beim Lambert-Eaton-Syndrom wird unter anderem nach Autoantikörpern gegen spannungsabhängige Calcium-Kanäle (VGCC-Antikörper) gesucht.
Beim kongenitalen myasthenen Syndrom werden spezielle molekulargenetische Untersuchungen durchgeführt. Dabei sucht man in den Zellen nach bestimmten Veränderungen des Erbmaterials (DNA).
- Pharmakologische Testung
In bestimmten Fällen (zum Beispiel bei der seronegativen MG, bei welcher keine spezifischen Antikörper nachgewiesen werden konnten) kommen AChE-Hemmer (AChE-Inhibitoren = AChE-I) unter ärztlicher Kontrolle zur Anwendung. Führt die Gabe des Medikaments zu einer Verbesserung der Symptome, erhärtet sich der Verdacht auf eine Myasthenia gravis.
- Bildgebende Verfahren
Zur Feststellung einer Erkrankung des Thymus sollten bildgebende Verfahren wie CT- oder MRT-Untersuchungen erfolgen. Bei der rein okulären Myasthenia gravis ohne Nachweis von Antikörpern können bildgebende Verfahren hilfreich sein, um lokale Prozesse, welche Veränderungen an der betroffenen Muskulatur bedingen, auszuschließen.
Therapie: Wie wird eine Myasthenie behandelt?
Myasthenie ist nicht heilbar. Sie ist aber meist sehr gut medikamentös unter Kontrolle zu bringen. Therapieziel ist es daher, die Lebensqualität der Betroffenen wiederherzustellen oder zumindest deutlich zu verbessern.
Dabei lässt sich die Krankheitskontrolle in vier Stufen einteilen:
- Volle Krankheitskontrolle und Beschwerdefreiheit
- Volle Krankheitskontrolle mit geringen Restbeschwerden
- Unvollständige Krankheitskontrolle mit wechselnd intensiven Beschwerden. Es kann immer wieder zu Krankheitsverschlechterungen (Krisen) kommen.
- Keine Krankheitskontrolle mit unverändertem Beschwerdebild und/oder zusätzlicher Verschlechterung.
Die Therapie muss an die individuelle Krankengeschichte der Betroffenen angepasst sein. Dabei richtet sich die Entscheidung einerseits nach dem Vorhandensein von Antikörpern. Andererseits ist das Beurteilen des Krankheitsverlaufs in den vergangenen Jahren immer wichtiger geworden. Unterteilt werden zwei Verlaufsformen:
- milder/moderater Verlauf
- aktiver/hochaktiver Verlauf
Wichtige Bausteine der Therapie sind:
- Symptomatische Therapie
- Verlaufsmodifizierende (Immun-)Therapie
Symptomatische Therapie
Hierunter versteht man eine Therapieform, welche die Beschwerden lindert, ohne die Ursache zu beseitigen. Hierzu wird meist ein AChE-I verwendet.
- Acetylcholinesterase-Hemmer (AChE-I)
Beim Vorhandensein von Autoantikörpern gegen den AchR kommen AChE-I zum Einsatz.
AChE ist das Enzym, welches im synaptischen Spalt den Botenstoff ACh abbaut (siehe Bildergalerie). Die Medikamente hemmen die AChE, sodass weniger ACh abgebaut werden kann und der Neurotransmitter damit länger im synaptischen Spalt verbleibt. Empfohlen wird der AChE-I Pyridostigmin.
AChE-I sind beim Lambert-Eaton-Syndrom weniger wirksam als bei einer Myasthenie. Mittel der Wahl ist hier eine Kombinationstherapie mit Amifampridin oder 3,4-Diaminopyridin (DAP) sowie Pyridostigmin.

Bei Myasthenie hakt die Signalübertragung am Muskel (links). Medikamente blockieren den Abbau des Signalstoffs ACh. Seine Konzentration erhöht sich, die Signalübertragung verbessert sich (rechtes Bild).
© W&B/Martina Ibelherr
Dieser Behandlungsansatz entspricht einer „symptomatischen Therapie“, da durch diesen Mechanismus die Beschwerden gelindert werden, aber nicht die Ursache behandelt wird. Als „ursächliche Therapie“ - das heißt zur Behandlung des Auslösers „Antikörper gegen körpereigene Strukturen“ - kommen folgende Medikamente/Ansätze infrage:
Verlaufsmodifizierende (Immun-) Therapie
- Immunsuppressiva
Medikamente, die das Abwehrsystem unterdrücken, heißen Immunsuppressiva und spielen bei der Behandlung der Myasthenie eine wichtige Rolle. Sie können verhindern, dass das Abwehrsystem Antikörper bildet. Meist kommt dabei eine Kombination von Glucocorticosteroiden mit Azathioprin zum Einsatz. Glucocorticosteroide sind häufig unter dem Begriff „Cortison-Therapie“ bekannt, Medikamente dieser Gruppe sind beispielsweise Prednison, Prednisolon und Methylprednisolon. Ist ein stabiler Gesundheitszustand erreicht, kann nach einiger Zeit versucht werden, die Medikamente abzusetzen. Dies darf nie plötzlich erfolgen, sondern nur in engmaschiger Absprache mit dem Arzt oder der Ärztin durch eine langsame Reduzierung der Dosis (ausschleichend).
Sollten oben genannte Medikamente unwirksam sein, kann auch die Behandlung mit anderen Präparaten wie Mycophenolat-Mofetil (MMF), Ciclosporin A (CSA), Tacrolimus oder Methotrexat (MTX) versucht werden.
- Weitere Medikamente
Für bestimmte Formen der Myasthenie können auch Komplementinhibitoren (Ravulizumab, Eculizumab) oder Modulatoren des neonatalen Fc-Rezeptors (FcRn) (Efgartigimod) in Betracht gezogen werden. Auch der Antikörper Rituximab kann bei schwereren, hochaktiven Verlaufsformen (insbesondere bei der anti-MuSK seropositiven MG) zum Einsatz kommen.
Operative Therapie
Ist eine Erkrankung des Thymus (Thymom, Thymitis) ursächlich für die Myasthenia gravis, sollte eine operative Entfernung der Thymusdrüse erfolgen. Vor allem bei Patient:innen im Alter zwischen 18 und 65 Jahren mit Autoantikörpern gegen den AChR wird der Eingriff (Thymektomie) empfohlen. Bei der seronegativen (kein Vorhandensein von Antikörpern) und der LRP-4 positiven Form kann die Thymektomie erwogen werden, insbesondere bei hoher Krankheitsaktivität. Handelt es sich um die MuSK-Antikörper positive Form wird der operative Eingriff aktuell nicht empfohlen.
Intensivmedizinische Behandlung
Im Falle einer myasthenen Krise ist eine Intensivtherapie, oftmals mit künstlicher Beatmung notwendig. Zusätzlich werden Verfahren wie die Plasmapherese (eine Art "Blutwäsche") oder Immunadsorption angewandt, um MG-spezifische Antikörper aus dem Blut zu entfernen. Auch eine hochdosierte Behandlung mit speziellen Antikörpern (intravenöse IgG-Antikörper, IvIg) kann wirksam sein, um die körpereigene Immunantwort zu verbessern.
Weitere Therapieoptionen
Bei der okulären Form der Myasthenia gravis, welche sich unter medikamentöser Therapie nicht ausreichend bessert, können auch mechanische Hilfsmittel (Lidretraktor an der Brille, durchsichtiges Klebeband) oder operative Maßnahmen wie eine Lidraffung zur Korrektur des herabhängenden Augenlids hervorragende Ergebnisse erzielen.
Ebenfalls gilt es mittlerweile als erwiesen, dass sich Bewegung positiv auf den Krankheitsverlauf auswirkt und eine vergrößerte Muskelkraft zu einer besseren Lebensqualität führt. Neben Bewegungs- und Trainingstherapien können auch ergänzende Maßnahmen, wie Physio- oder Ergotherapie, Logopädie, neuropsychologische Therapie und Entspannungsmaßnahmen zu einem positiven Krankheitsverlauf beitragen.
Auch Selbsthilfeorganisationen können Betroffene unterstützen, zu nennen ist hier insbesondere die Deutsche Myasthenie-Gesellschaft
Was müssen Menschen mit einer Myasthenie beachten?
Es gibt zahlreiche Medikamente, die die Symptome einer Myasthenie verstärken können. Dazu gehören einige Antibiotika wie:
- Makrolide (wie Telithromycin, Erythromycin, Azithromycin, Clarithromycin)
- Fluorchinolone (wie Ciprofloxacin, Levofloxacin, Norfloxacin)
- Aminoglykoside (wie Gentamicin, Neomycin, Tobramycin)
Auch Mittel zur Blockade der neuromuskulären Übertragung wie Botulinumtoxin und Betablocker (über die Vene verabreicht) können eine Myasthenie verschlechtern. Bestimmte Mittel in der Anästhesie können ebenfalls die neuromuskuläre Übertragung verschlechtern.
Es ist wichtig, dass die Betroffenen über diese Zusammenhänge informiert sind, um Komplikationen zu vermeiden.

Professor Heinz Wiendl
© W&B/Privat
Unser beratender Experte:
Professor Dr. med. Heinz Wiendl ist Direktor der Neurologischen Klinik mit Institut für Translationale Neurologie der Universität Münster. Sein Forschungsschwerpunkt ist unter anderem die funktionelle Interaktion des Immunsystems mit dem Nervensystem. Er ist der federführende Verfasser der Leitlinien zur Diagnostik und Therapie der Myasthenia Gravis und des Lambert-Eaton-Syndroms. Er ist Mitglied in verschiedenen Fachgesellschaften, darunter der Ärztliche Beirat der deutschen Multiple Sklerose Gesellschaft.
Wichtiger Hinweis:
Dieser Artikel enthält nur allgemeine Hinweise und darf nicht zur Selbstdiagnose oder –behandlung verwendet werden. Er kann einen Arztbesuch nicht ersetzen. Die Beantwortung individueller Fragen durch unsere Experten ist leider nicht möglich.