

Eiweiß-Origami
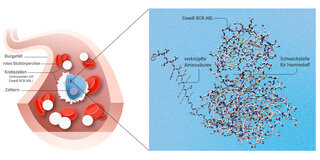
© Cowan-Jacob S.W. et al. Acta Crystallogr D Biol Crystallogr (2007) 63: 80-93/bearb. Infografik: W&B/Michelle Günther

Warum wird jetzt darüber geredet?
„Es war der Traum über Jahrzehnte. Dass er sich so schnell erfüllt hat, war für die meisten von uns eine Überraschung.“ So kommentiert Professor Markus Lill vom Departement Pharmazeutische Wissenschaften an der Universität Basel (Schweiz) das Ergebnis eines wissenschaftlichen Wettbewerbs vor zwei Jahren. Es ging darum, den räumlichen Aufbau von Eiweißen möglichst exakt vorherzusagen.
Ein bestimmtes Computerprogramm schlug seine Konkurrenten um Längen. Es errechnete in wenigen Stunden 3-D-Strukturen von Eiweißen bis in ihren atomaren Aufbau hinein mit großer Genauigkeit. Die Ergebnisse waren genauso gut wie die von experimentellen Strukturanalysen. Dazu werden Eiweiße kristallisiert und mit Röntgenstrahlen durchleuchtet. Oder sie werden tiefgefroren und dann mit Elektronenmikroskopen untersucht.
Bereits im Jahr 1958 wurde die erste 3-D-Struktur experimentell entschlüsselt: die von Myoglobin, dem sauerstoffbindenden Eiweiß in Muskeln. Doch solche Laboranalysen dauern mitunter Jahre, und sie führen längst nicht bei jedem untersuchten Eiweiß zum Erfolg.
Warum klappte das nicht schon früher?
Jedes Eiweiß entsteht zunächst als Kette aneinandergeknüpfter Aminosäuren. Im Bruchteil einer Sekunde falten sie sich von selbst zu einem dreidimensionalen Gebilde, das seine Aufgabe im Körper erfüllt: zum Beispiel Sauerstoff binden. Seit etwa fünfzig Jahren versuchen Forscherinnen und Forscher zu errechnen, was die Natur von allein schafft.
Doch die Anzahl der prinzipiell möglichen Faltungen ist nahezu grenzenlos: „Es würde länger als das Alter des bekannten Universums dauern, um alle möglichen Konfigurationen eines typischen Eiweißes durchzuspielen“, sagte der US-Molekularbiologe Cyrus Levinthal im Jahr 1969. Eine unfassbar große Zahl mit dreihundert Nullen.
Die Künstliche Intelligenz schaffte den Durchbruch. Das Programm lernte anhand bekannter Strukturen Regeln für die Faltung und übertrug sie auf andere Eiweiße – darunter solche mit noch unbekanntem Aufbau. „Ein unglaublich kompliziertes Netzwerk von Berechnungen wurde trainiert, Milliarden von Einflussgrößen optimiert“, sagt Professor Gunnar Schröder vom Institut für Biologische Informationsprozesse des Forschungszentrums Jülich.
Was haben Kranke davon?
Viele Medikamente fußen unter anderem auf Strukturinformationen ihrer Angriffsziele. Etwa die Substanz Imatinib gegen Blutkrebs. Das Computerprogramm könnte die Entdeckung von Wirkstoffen generelleinfacher und schneller machen. Mehr als 214 Millionen 3-D-Modelle hat es inzwischen errechnet, also den Aufbau fast aller Eiweiße sämtlicher untersuchten Arten: Vom Hefepilz über Krankheitserreger biszum Menschen. 35 Prozent treffen so präzise zu wie experimentell bestimmte Strukturen. Weitere 45 Prozent sind recht genau.

„Allerdings ist noch nicht klar, wie gut die Modelle jetzt schon für die Medikamentenforschung taugen“, räumt Markus Lill ein. Denn fast alle Wirkstoffe binden an flexible Abschnitte von Eiweißen. Diese lassen sich weniger genau errechnen als die starren Bereiche. Doch viele Experimentatoren seien von den exakten Prognosen schockiert gewesen. Ob Computer künftig die Laborarbeit ersetzen, werden die kommenden Jahre zeigen.
Quellen:
- Kendrew, J.C., Bodo, G., Dintzis, H. et al.: A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. In: Nature 01.01.1958, 181: 662-666
- Cyrus Levinthal : How to fold graciously. In: Mössbaun Spectroscopy in Biological Systems Proceedings.Univ. of Illinois Bulletin 01.01.1969, 67: 22-24
- Cowan-Jacob SW, Fendrich G, Floersheimer A, et al.: Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. In: Acta Crystallogr D Biol Crystallogr 01.01.2007, 63: 80-93
- Jumper J, Evans R, Pritzel A, et al. : Highly accurate protein structure prediction with AlphaFold. In: Nature 01.01.2021, 596-7873: 583-589
- Tunyasuvunakool K, Adler J, Wu Z et al.: Highly accurate protein structure prediction for the human proteome. In: Nature 01.01.2021, 596-7873: 590-596
- Baek M, DiMaio F, Anishchenko I et al. : Accurate prediction of protein structures and interactions using a three-track neural network. In: Science 01.01.2021, 373-6557: 871-876
- Callaway E: 'The entire protein universe': AI predicts shape of nearly every known protein. In: Nature 28.07.2022, 608: 15-16