
zum Artikel














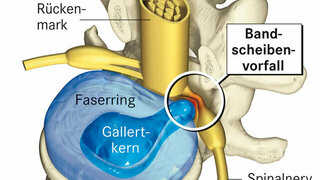






Haben Sie gewonnen?
Hier finden Sie alle Preisrätsel-Gewinner aus der "Apotheken Umschau"








Die Anzeige der externen Inhalte wurde von Ihnen im Consent abgewählt.
Wollen Sie die Inhalte doch angezeigt bekommen?




